

Epigenomics
Epigenomics
Science Highlights
Successes of a Comprehensive Consortium in Human Epigenomics
For the Public
Scientists have long been aware about the importance of our DNA; it is the blueprint for all the vital components of our cells to keep us healthy. Over time, we have learned that DNA is a dynamic molecule and that it, along with its associated proteins, can be chemically modified to change how the blueprint is read and used by our cells. These are referred to as epigenomic changes. While scientists have been rapidly learning more about these changes recently, when the NIH Common Fund’s Epigenomics program started in 2008, there was a great need for information and tools to help researchers study epigenomics in human cells and tissues.
The Epigenomics program generated a set of reference epigenomes and new research tools, technologies, datasets, and infrastructure that accelerated our understanding of how epigenomic modifications to DNA regulate gene activity. These research resources also expanded our awareness regarding the role these modifications play in health and disease. More information about the success of the Epigenomics program can be found in a Science Advances article which summarizes the entirety of the Epigenomics program.
For Researchers
There are many types of epigenomic modifications and their occurrence and biological significance across the human genome has not been well-characterized. In 2008, the NIH launched an Epigenomics program, and one of its goals was mapping modifications across the entire genome of many different human cells and tissue types. These epigenomic maps are available to the entire scientific community for use in their own research projects through the National Center for Biotechnology Information at the NIH (https://www.ncbi.nlm.nih.gov/geo/roadmap/epigenomics/). More can be learned about the Epigenomics mapping effort, including the relevant protocols used to obtain data and the numerous publications generated by the mapping consortium on the Common Fund Roadmap Epigenomics program website.
In addition to generating epigenomic reference maps, the Epigenomics program developed various databases, tools, and resources for the research community; both by generating them internally as well as through establishing international and domestic collaborations. The Epigenomic datasets are available through the Roadmap Epigenomics Mapping Consortium website, the ENCODE website, IHEC, and the Baylor Epigenome Atlas. Over 230 publications, covering a wide range of topics, have cited use of Roadmap Epigenomics program data. The Roadmap Epigenomics program became a founding member of the International Human Epigenome Consortium (IHEC) in 2010, which has significantly enhanced global funding and coordination of epigenomic research. More information about the success of the Epigenomics program can be found in this Science Advances article which summarizes the entirety of the Epigenomics program.
Reference:
The NIH Common Fund/Roadmap Epigenomics Program: Successes of a Comprehensive Consortium. John Satterlee, Lisa Chadwick, Frederick Tyson, Kim McAllister, Jill Beaver, Linda Birnbaum, Nora Volkow, Elizabeth Wilder, James Anderson, and Ananda Roy. Science Advances, Vol 5 (7), Jul 2019, DOI: 10.1126/sciadv.aaw6507
 X-Linked Disorders: When One Healthy Gene Isn't Enough
X-Linked Disorders: When One Healthy Gene Isn't Enough
Specialized human chromosomes called the X chromosome and Y chromosome determine our sex. While males have one X and  one Y chromosome, females have two X chromosomes. The X chromosome contains hundreds of genes that are associated with diseases. Most of these diseases are more common in males because they only have one X chromosome, so inheriting just one faulty copy of an X-linked gene can result in the disease. However, some of these diseases also occur in females, even if they only inherit one faulty copy of the gene. This is because one of the X chromosomes in each female cell is inactive. Inactivation results, in part, from high levels of DNA modification (methylation) that prevent gene activity, along with the production of an inhibitory molecule called long non-coding RNA(named Xist) that binds to the X chromosome. If the protein produced from a gene is critical for cell function, having the faulty gene active in some of the cells results in disease. There are currently no disease-specific treatments for this group of disorders. However, an emerging strategy for treating X-linked diseases in females is reactivation of the X chromosome containing the healthy version of the gene.
one Y chromosome, females have two X chromosomes. The X chromosome contains hundreds of genes that are associated with diseases. Most of these diseases are more common in males because they only have one X chromosome, so inheriting just one faulty copy of an X-linked gene can result in the disease. However, some of these diseases also occur in females, even if they only inherit one faulty copy of the gene. This is because one of the X chromosomes in each female cell is inactive. Inactivation results, in part, from high levels of DNA modification (methylation) that prevent gene activity, along with the production of an inhibitory molecule called long non-coding RNA(named Xist) that binds to the X chromosome. If the protein produced from a gene is critical for cell function, having the faulty gene active in some of the cells results in disease. There are currently no disease-specific treatments for this group of disorders. However, an emerging strategy for treating X-linked diseases in females is reactivation of the X chromosome containing the healthy version of the gene.
NIH Roadmap Epigenomics program investigator Dr. Jeannie Lee led a research team to explore a new method of X reactivation that combines two approaches – inhibiting DNA methylation and simultaneously interfering with the RNA responsible for X inactivation (Xist). The team tested this approach in mouse cells to treat Rett Syndrome, which results from the mutation of a gene on the X chromosome that codes for the protein “MECP2.” MECP2 is important for development of nerve cells. Using this approach, the researchers saw 30,000 times greater production of MECP2 due to reactivation of the inactive X chromosome containing the healthy gene. The next step will be to test this treatment approach in a Rett Disorder animal model. The results of this study indicate that this new mixed approach is a promising treatment strategy for some X-linked disorders.
Reference: A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Carrette, LL, Wang, CY, Wei, C, Press, W, Ma, W, Kelleher, RJ, and Lee, JT. PNAS. 2017 Dec 27. 115(4):E668-E675.
CRISPR Epigenome Editing Hits the Big Time
 The human genome can remarkably transform intricate biological signals into complex patterns of gene expression (the process by which genes are first converted to RNA and then to proteins). Differences in gene expression is what gives individuals unique characteristics. Gene expression is controlled by regulatory elements, segments of noncoding DNA (that does not encode protein) which are capable of increasing or decreasing the expression of a targeted gene. Although noncoding genetic variation is associated with disease (including cancer and cardiovascular disease), there are many unanswered questions about how these noncoding regulatory elements work; even identifying them can be a major challenge. Efforts like the ENCODE Project and the NIH Common Fund Roadmap Epigenomics Program have aimed to address this challenge by identifying millions of putative regulatory elements, across over a hundred different human cell types. Yet, the function of these regulatory elements remains largely unknown.
The human genome can remarkably transform intricate biological signals into complex patterns of gene expression (the process by which genes are first converted to RNA and then to proteins). Differences in gene expression is what gives individuals unique characteristics. Gene expression is controlled by regulatory elements, segments of noncoding DNA (that does not encode protein) which are capable of increasing or decreasing the expression of a targeted gene. Although noncoding genetic variation is associated with disease (including cancer and cardiovascular disease), there are many unanswered questions about how these noncoding regulatory elements work; even identifying them can be a major challenge. Efforts like the ENCODE Project and the NIH Common Fund Roadmap Epigenomics Program have aimed to address this challenge by identifying millions of putative regulatory elements, across over a hundred different human cell types. Yet, the function of these regulatory elements remains largely unknown.
What makes studying regulatory elements so difficult? In addition to difficulties in determining which genes they target, the sheer number of putative regulatory elements that have been identified (millions) presents a major hurdle. Existing approaches use high throughput methods – essentially carrying out thousands of experiments at once – but these methods can disrupt the natural or “native” state of the genome. Further, these methods monitor activity of a regulatory element, but cannot identify the gene they target. CRISPR is another tool which uses genomic engineering to target specific regions of the genome and edit DNA at precise locations, commonly for inactivating or “turning off” genes. Although CRISPR can be used to study regulatory elements, using it to edit genomic DNA alone is not ideal due to the complexity of regulatory element interactions with cellular machinery.
To overcome these hurdles, Common Fund Roadmap Epigenomics Program grantee Dr. Charles Gersbach, Associate Professor of Biomedical Engineering at Duke University, and collaborators, have developed a CRISPR-based epigenomic regulatory element screening (CERES) approach for improved high throughput screening of regulatory elements in their native context. By combining CRISPR with epigenome modifying proteins, they were able to carry our screens that both inactivated and activated genes via modifications “on top of” the genome. Researchers also identified the regulatory elements for the beta-globin and HER2 genes in human cells. Beta-globin is makes up the most common form of hemoglobin in adult humans, while HER2 is a gene that can play a role in the development of breast cancer. This powerful epigenome editing tool can be used to help researchers understand the function of regulatory elements and how they coordinate gene expression, which will help us understand the role of altered gene expression in human health and disease.
Reference: CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, Crawford GE, Reddy TE, Gersbach CA. Nature Biotechnology. 2017 April 3.
In the News: CRISPR Screen Detects Functional Gene Regulation, The Scientist. Screening the Human Genome’s ‘Dark Matter’, Duke University.
International Human Epigenome Consortium (IHEC) Celebrates Major Coordinated Paper Release
 Investigators from the International Human Epigenome Consortium (IHEC), which includes Common Fund Roadmap Epigenomics Program grantees, have published a special collection of papers titled “Insights from the International Human Epigenome Consortium”. This collection includes 24 papers published in Cell and other Cell Press journals, plus 17 papers published elsewhere. This publication release demonstrates how the consortium’s epigenomic reference maps can help find answers to pressing questions related to the cellular mechanisms associated with numerous complex human diseases. For example, Common Fund Epigenomics grantee Dr. Aleksandar Milosavljevic and collaborators found that the epigenetic profiles of breast cancer cells are distinct from normal epithelial cells and that these epigenetic marks can be mapped to specific groups of cancer cells. This is significant because it enables the study of factors that drive tumor progression.
Investigators from the International Human Epigenome Consortium (IHEC), which includes Common Fund Roadmap Epigenomics Program grantees, have published a special collection of papers titled “Insights from the International Human Epigenome Consortium”. This collection includes 24 papers published in Cell and other Cell Press journals, plus 17 papers published elsewhere. This publication release demonstrates how the consortium’s epigenomic reference maps can help find answers to pressing questions related to the cellular mechanisms associated with numerous complex human diseases. For example, Common Fund Epigenomics grantee Dr. Aleksandar Milosavljevic and collaborators found that the epigenetic profiles of breast cancer cells are distinct from normal epithelial cells and that these epigenetic marks can be mapped to specific groups of cancer cells. This is significant because it enables the study of factors that drive tumor progression.
View the full collection of datasets and analyses here and read the full story here.
In the news: More than 40 New Papers on Epigenetics Published, The Scientist.
Epigenetic Imaging Shows Gene Activation in Living Brains for the First Time
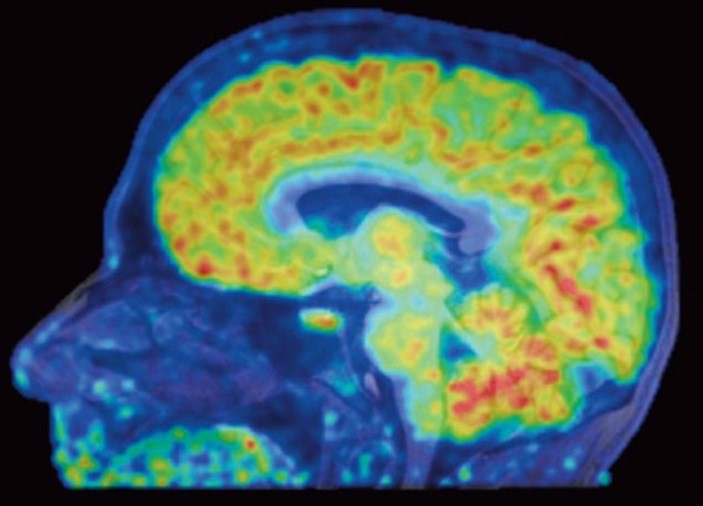 Epigenomics grantee Dr. Jacob Hooker and collaborators have used a new neuroimaging tool to show, for the first time, where genes are being turned off or on in living human brains. Histone deacetylases (HDACs) are enzymes that regulate gene expression through epigenetic modifications and are therefore useful therapeutic targets. Using a specific HDAC imaging probe called Martinostat and positron emission tomography (PET) scanning, Dr. Hooker’s group has visualized HDAC expression in the living brain of eight healthy volunteers. In addition to observing distinct regions of HDAC expression within human brains regions, they also saw strikingly conserved regions of HDAC expression levels between these individuals. These conserved patterns within and between healthy individuals are significant because this lays the groundwork for understanding epigenetic information in the human central nervous system (CNS) and related diseases. “I’m hoping these colorful maps let us compare healthy brains with the brains of people with schizophrenia, Alzheimer’s, and other diseases,” said Hooker. The authors conclude that this work provides a “critical foundation for how to quantify epigenetic activity in the living brain and in turn accomplish HDAC inhibition in the CNS as a therapy for human brain disorders”.
Epigenomics grantee Dr. Jacob Hooker and collaborators have used a new neuroimaging tool to show, for the first time, where genes are being turned off or on in living human brains. Histone deacetylases (HDACs) are enzymes that regulate gene expression through epigenetic modifications and are therefore useful therapeutic targets. Using a specific HDAC imaging probe called Martinostat and positron emission tomography (PET) scanning, Dr. Hooker’s group has visualized HDAC expression in the living brain of eight healthy volunteers. In addition to observing distinct regions of HDAC expression within human brains regions, they also saw strikingly conserved regions of HDAC expression levels between these individuals. These conserved patterns within and between healthy individuals are significant because this lays the groundwork for understanding epigenetic information in the human central nervous system (CNS) and related diseases. “I’m hoping these colorful maps let us compare healthy brains with the brains of people with schizophrenia, Alzheimer’s, and other diseases,” said Hooker. The authors conclude that this work provides a “critical foundation for how to quantify epigenetic activity in the living brain and in turn accomplish HDAC inhibition in the CNS as a therapy for human brain disorders”.
In the news: In living color: New technique sees gene activity in human brains, STAT News. Also featured on PBS Newshour and Scientific American.
Reference: Insights into neuroepigenetics through human histone deacetylase PET imaging. Wey HY, Gilbert TM, Zürcher NR, She A, Bhanot A, Taillon BD, Schroeder FA, Wang C, Haggarty SJ, Hooker JM. Science Translational Medicine. 2016 August. 8(351):351ra196. PMID: 27510902.
Precise Epigenetic Control of Gene Expression Using CRISPR
 Researchers supported by the NIH Roadmap Epigenomics Program have demonstrated a method to use the CRISPR system to switch the human epigenome on or off at specific positions, without altering the underlying genetic code. Originally employed to “knock out” genes, the CRISPR system uses a guide RNA that targets a specific DNA sequence and a Cas9 nuclease that “snips” this DNA. This system can be adapted for epigenome editing, if the Cas9 nuclease is inactivated and teamed with a protein that controls the epigenetic packaging of the genome. Whether used for genomic or epigenomic manipulation, CRISPR is not always precise. Evaluating off-target effects and specificity are important steps in assessing the utility of CRISPR technology for studying human disease.
Researchers supported by the NIH Roadmap Epigenomics Program have demonstrated a method to use the CRISPR system to switch the human epigenome on or off at specific positions, without altering the underlying genetic code. Originally employed to “knock out” genes, the CRISPR system uses a guide RNA that targets a specific DNA sequence and a Cas9 nuclease that “snips” this DNA. This system can be adapted for epigenome editing, if the Cas9 nuclease is inactivated and teamed with a protein that controls the epigenetic packaging of the genome. Whether used for genomic or epigenomic manipulation, CRISPR is not always precise. Evaluating off-target effects and specificity are important steps in assessing the utility of CRISPR technology for studying human disease.
To investigate the specificity of CRISPR tools in the context of epigenomic editing, Dr. Charles Gersbach, Ph.D., a professor of biomedical engineering at Duke University, worked with a team of scientists to conduct gene-silencing experiments. For these studies, an inactive Cas9 nuclease was teamed with a Krüppel-associated box (KRAB) transcriptional repressor protein. They then measured the degree to which Cas9-KRAB demonstrated genome-wide specificity, as well as the extent to which it catalyzed epigenomic packaging. The authors show that repression mediated by Cas9-KRAB is sufficiently specific to disrupt gene expression via modification of the epigenome at specific positions.
“The primary advantage of CRISPR over previous technologies is the ability to use a genetic scalpel rather than a sledgehammer,” said Dr. Gersbach. “Many labs across the world are using these tools on the assumption that they’re getting specific effects, but there had not been a comprehensive analysis to prove it. These experiments show exceptional specificity, demonstrating that the technology is capable of targeting single sequences of the genome.” For additional information, read the Duke University News Release.
Reference:Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Thakore PI, D’Ippoltio AM, Song L, Safi A, Shivakumar NK, Kadabi AM, Reddy TE, Crawford GE, Gersbach CA. Nature Methods. 2015 December. 12(12): 1143-49. PMID: 4666778.
Uncovering Important Epigenetic Changes in Neuronal Cell Development
 Duke Researchers funded by the Common Fund are utilizing epigenomics to enrich our understanding of development of the human brain. With increased understanding of how and when epigenetic marks are influencing gene expression, researchers may be able to predict and understand more about the fate of particular neurons and understand fundamental principles of gene regulation in the brain. Highly comprehensive epigenomic data generated from the West lab at Duke University is uncovering more information about the extremely dynamic system coordinating the intricate temporal pattern of neuronal development.
Duke Researchers funded by the Common Fund are utilizing epigenomics to enrich our understanding of development of the human brain. With increased understanding of how and when epigenetic marks are influencing gene expression, researchers may be able to predict and understand more about the fate of particular neurons and understand fundamental principles of gene regulation in the brain. Highly comprehensive epigenomic data generated from the West lab at Duke University is uncovering more information about the extremely dynamic system coordinating the intricate temporal pattern of neuronal development.
The study uses a technique called DNase I Hypersensitivity (DHS) to map chromatin accessibility - sites in the genome that are structurally accessible to the cellular machinery that turn on and off genes- in different stages of neuron development. Using the developing mouse cerebellar cortex that is primarily comprised of a single type of neuron, a cerebellar granule neuron (CGN), they were able to study a highly specific cell population. They mapped chromatin accessibility in these cells at three key times in postnatal development and found highly dynamic changes at over 20,000 regulatory sites during these different stages. Upon further analysis many of these enhancer sites contained binding sites for several transcription factor families; including zinc finger proteins of the cerebellum (ZIC). Following up with functional studies, the researchers confirmed a previously unknown role for these ZICs in coordinating gene expression in the growth and maturation of neurons. Furthermore they found that marks remained even after a gene was turned off—labeling the genes for an easy “start up” later. This raises the possibility that the accessibility of enhancer sites can be in a “primed” state which has implications for learning and memory. Ultimately, understanding more about these the epigenetic changes in neuronal cell development across the genome may help researchers understand and potentially design therapies targeting diseases of the brain.
span style="font-size: 12px">Reference:![]() Regulation of Chromatin Accessibility and Zic Binding at Enhancers in the Developing Cerebellum. Frank, C. L., F. Liu, R. Wijayatunge, L. Song, M. T. Biegler, M. G. Yang, C. M. Vockley, A. Safi, C. A. Gersbach, G. E. Crawford, and A. E. West. Nature Neuroscience. 2015 May.18(5):647-56. PMID: 25849986.
Regulation of Chromatin Accessibility and Zic Binding at Enhancers in the Developing Cerebellum. Frank, C. L., F. Liu, R. Wijayatunge, L. Song, M. T. Biegler, M. G. Yang, C. M. Vockley, A. Safi, C. A. Gersbach, G. E. Crawford, and A. E. West. Nature Neuroscience. 2015 May.18(5):647-56. PMID: 25849986.
Common Fund-Supported Researchers Develop Powerful Tools to Edit Genomes
 Epigenetic marks are chemical modifications to DNA or DNA-associated proteins that regulate gene expression without changing the DNA sequence. These marks act in many important processes including development, aging, health, and disease; because of this they are targets for therapeutic intervention and intense research activity. Exciting new technologies, such as the CRISPR/Cas9 system used for gene editing (adding, deleting or changing the sequence of targeted genes), have opened new possibilities and sparked a transformation in genetic and epigenomic research (the study of epigenetics across the full genome). Researchers that are funded through the Common Fund’s Epigenomics Program and New Innovator Award at Dr. Gersbach’s lab at Duke University have recently exploited CRISPR/Cas9 technologies to develop state-of-the-art tools that are shaping new discoveries.
Epigenetic marks are chemical modifications to DNA or DNA-associated proteins that regulate gene expression without changing the DNA sequence. These marks act in many important processes including development, aging, health, and disease; because of this they are targets for therapeutic intervention and intense research activity. Exciting new technologies, such as the CRISPR/Cas9 system used for gene editing (adding, deleting or changing the sequence of targeted genes), have opened new possibilities and sparked a transformation in genetic and epigenomic research (the study of epigenetics across the full genome). Researchers that are funded through the Common Fund’s Epigenomics Program and New Innovator Award at Dr. Gersbach’s lab at Duke University have recently exploited CRISPR/Cas9 technologies to develop state-of-the-art tools that are shaping new discoveries.
Using the powerful CRISPR/Cas9 system, Dr. Gersbach and colleagues made a fusion protein that activates particular genes by directly targeting specific histones for chemical changes, such as acetylation-one type of epigenetic mark. Currently, studying the function of particular epigenetic marks has largely been limited to statistical associations with gene expression patterns, and not to direct functional studies. This unique molecular tool allows researchers to surmount this challenge. With this system, the group showed this directed acetylation to promoter and enhancer regions is sufficient to turn on gene expression. This novel system provides a powerful tool for researchers to directly turn on or off targeted genes of interest thereby allowing direct functional analysis of site-specific epigenetic modifications.
In another innovative application of CRISPR-Cas9 technology, Dr. Gersbach and colleagues were able to control gene expression by simply turning light on or off. They developed a system where they made two fusion proteins, fusion proteins are made by the joining of two or more genes that originally coded for separate proteins. One protein consisted of an inactivated Cas9 protein fused with a plant protein called CIB1 and the other protein fused a transcriptional activation domain to cryptochrome 2 (CRY2). When both proteins and a guide RNA are present in cells and illuminated with blue light, the two fusion proteins pair up, bind to their DNA, and turn on gene expression. Future application of this innovative technique could allow researchers to target any gene in an organism and turn it on or off with the switch of a light, which has amazing potential to transform genetic engineering.
References:
Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Hilton, I. B., A. M. D'Ippolito, C. M. Vockley, P. I. Thakore, G. E. Crawford, T. E. Reddy, and C. A. Gersbach. Nature Biotechnology. 2015 May. 33(5): 510-7. PMID: 25849900.
A Light-Inducible CRISPR-Cas9 System for Control Of Endogenous Gene Activation. Polstein, L. R., and C. A. Gersbach. Nature Chemical Biology. 2015 March. 11(3) 198-200. PMID: 25664691.
Common Fund-supported Researchers Map Epigenome of More Than 100 Tissue and Cell Types
 More than 440 researchers in 32 labs around the world participating in the Common Fund’s Roadmap Epigenomics program have generated and analyzed 111 reference human epigenomes. This is the largest collection to date of reference human epigenomes from a broad range of representative primary cells and tissues. A genome is defined as the DNA sequences present in a cell, and an epigenome refers to the chemical modifications and non-sequence changes to DNA and DNA-associated proteins. Currently, researchers are beginning to understand the many consequences of these chemical modifications, and early insights from this new analysis reveal that different epigenomic states are associated with differences in age, sex, and tissue type. The many companion papers describing additional analyses demonstrate the broad applicability of the resource. These studies examine the biological importance of epigenetic changes in the context of stem cell differentiation, obesity, Alzheimer’s disease, cancer, and cardiac disease. This remarkable accomplishment will usher in new scientific understanding of the complexity of the human genome at a deeper level and spur new research advances in human health and disease.
More than 440 researchers in 32 labs around the world participating in the Common Fund’s Roadmap Epigenomics program have generated and analyzed 111 reference human epigenomes. This is the largest collection to date of reference human epigenomes from a broad range of representative primary cells and tissues. A genome is defined as the DNA sequences present in a cell, and an epigenome refers to the chemical modifications and non-sequence changes to DNA and DNA-associated proteins. Currently, researchers are beginning to understand the many consequences of these chemical modifications, and early insights from this new analysis reveal that different epigenomic states are associated with differences in age, sex, and tissue type. The many companion papers describing additional analyses demonstrate the broad applicability of the resource. These studies examine the biological importance of epigenetic changes in the context of stem cell differentiation, obesity, Alzheimer’s disease, cancer, and cardiac disease. This remarkable accomplishment will usher in new scientific understanding of the complexity of the human genome at a deeper level and spur new research advances in human health and disease.
Reference: Integrative analysis of 111 reference human epigenomes. Roadmap Epigenomics Consortium et al. Nature. 2015 Feb. 518(7539):317-330. PMID: 25693563.
Read the NIH press release here.
See video on the Epigenome by Nature here.
Double Meaning: Researchers Discover Hidden Codes in Genes
 Researchers in the Common Fund’s Epigenomics program have discovered a hidden layer of meaning contained within genes. Dr. John Stamatoyannopoulos at the University of Washington, along with his colleagues, have discovered some regions of DNA serve a double purpose. These regions contain instructions for how to make a protein, as well as information about when and how much of the protein should be made. Scientists previously thought that a particular stretch of DNA could be part of the genetic code, specifying the sequence of amino acid “building blocks” used to make a protein, or part of the regulatory code, containing elements that control expression of the protein. Dr. Stamatoyannopoulos and colleagues have identified “duons,” stretches of DNA within the genetic coding regions that also contain a regulatory sequence called a transcription factor binding site. The researchers created a map showing where transcription factors were bound within genetic coding regions. Looking across 81 diverse human cell types, they found that approximately 15 percent of DNA within genetic coding regions has this dual purpose. This study suggests that mutations within these duons could alter the protein sequence itself, the regulation of the protein, or possibly both simultaneously. These results have important implications for how researchers interpret genetic mutations to provide information about human health and disease.
Researchers in the Common Fund’s Epigenomics program have discovered a hidden layer of meaning contained within genes. Dr. John Stamatoyannopoulos at the University of Washington, along with his colleagues, have discovered some regions of DNA serve a double purpose. These regions contain instructions for how to make a protein, as well as information about when and how much of the protein should be made. Scientists previously thought that a particular stretch of DNA could be part of the genetic code, specifying the sequence of amino acid “building blocks” used to make a protein, or part of the regulatory code, containing elements that control expression of the protein. Dr. Stamatoyannopoulos and colleagues have identified “duons,” stretches of DNA within the genetic coding regions that also contain a regulatory sequence called a transcription factor binding site. The researchers created a map showing where transcription factors were bound within genetic coding regions. Looking across 81 diverse human cell types, they found that approximately 15 percent of DNA within genetic coding regions has this dual purpose. This study suggests that mutations within these duons could alter the protein sequence itself, the regulation of the protein, or possibly both simultaneously. These results have important implications for how researchers interpret genetic mutations to provide information about human health and disease.
Reference: Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynolds A, Raubitschek A, Ziegler S, LeProust EM, Akey JM, Stamatoyannopoulos JA. Exonic Transcription Factor Binding Directs Codon Choice and Affects Protein Evolution. Science, December 2013. 342; 1367-1372. PMID: 24337295.
Read the University of Washington press release here.
Cell Snapshot of Histone Marks
Epigenomics researcher Dr. Yingming Zhao has published a Cell Snapshot summarizing all known histone marks from human, mouse, and rat. Dr. Zhao's group discovered a significant proportion of the 400 currently known marks. Many histone marks play important roles in regulating gene expression, and identifying and understanding the specific functions of each of these marks will enable research into many aspects of human health and disease.
View the Cell Snapshot: Histone Modifications
Jumping genes have a job to do
 DNA is perceived to be a stable “blueprint” molecule since it encodes the proteins that function within a cell and also passes genetic information through the generations. However, DNA is actually extremely dynamic in many ways. Just one example of how DNA can be altered in surprising ways is by transposable elements (TEs), also called “jumping genes.” TEs, which make up approximately half the human genome, are sequences of DNA that move from one location in the genome to another. Scientists previously thought that TEs were silenced in the human genome, tagged with epigenetic marks to ensure that TEs are locked in place and prevented from disrupting the normal functions of the genome. However, it has recently become appreciated that in some cases, TEs play important roles in regulating gene expression. However, it is not well understood how this occurs or how widespread this phenomenon is.
DNA is perceived to be a stable “blueprint” molecule since it encodes the proteins that function within a cell and also passes genetic information through the generations. However, DNA is actually extremely dynamic in many ways. Just one example of how DNA can be altered in surprising ways is by transposable elements (TEs), also called “jumping genes.” TEs, which make up approximately half the human genome, are sequences of DNA that move from one location in the genome to another. Scientists previously thought that TEs were silenced in the human genome, tagged with epigenetic marks to ensure that TEs are locked in place and prevented from disrupting the normal functions of the genome. However, it has recently become appreciated that in some cases, TEs play important roles in regulating gene expression. However, it is not well understood how this occurs or how widespread this phenomenon is.
Drs. Ting Wang and Joseph Costello, supported by the Epigenomics program, along with their colleagues, identified epigenetic signatures marking regions of TEs that act as enhancers, regions of DNA that play a role in promoting gene expression. Intriguingly, the epigenetic signatures marking enhancer regions of TEs occur in a tissue-specific manner, and can be used to distinguish different tissues or possibly even cell types within tissues. The researchers also showed that these tissue-specific TE enhancer regions can influence expression of genes known to play important roles in the relevant tissue. For example, a TE region with an epigenomic signature for brain cells, but not immune cells, was found to influence expression of a gene involved in communication between brain cells, and did not influence expression of a gene involved in immune system responses. This research suggests that TEs may play a much more wide-spread role in tissue-specific gene regulation than was previously thought, and highlights the importance of including TEs in models of genetic and epigenetic regulation.
This research also used data from the ENCODE (ENCyclopedia Of DNA Elements) project at the National Human Genome Research Institute (NHGRI). Read more about ENCODE here.
Reference: Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, Gascard P, Sigaroudinia M, Tlsty TD, Kadlecek T, Weiss A, O’Geen H, Farnham JP, Madden PA, Mungall AJ, Tam A, Kamoh B, Cho S, Moore R, Hirst M, Marra MA, Costello JF, Wang T. DNA hypomethylation within specific transposable elements families associates with tissue-specific enhancer landscape. Nature Genetics, 2013, Jul. 45(7): 836-41. PMID: 23708189.
Common Fund Researchers Detail Epigenomic Changes during Development
 Most cells in the human body contain the same DNA, yet different types of cells have vastly different shapes, sizes, and functions. How do these differences arise? Chemical modifications to DNA and DNA-associated proteins, called epigenetic modifications, help instruct a cell to express only a sub-set of genes, giving rise to different characteristics for different cell types. Epigenetic regulation of gene expression changes during development, and can also change as a result of environmental exposures, pharmaceuticals, aging, and diet. Some epigenetic changes promote health and normal development, while others may contribute to a variety of diseases. Three recent publications in the journal Cell from the Epigenomics program’s Reference Epigenome Mapping Centers reveal important insights about epigenomic changes that take place during development, as non-specialized stem cells differentiate into specific cell types, such as heart, brain, skin, and many more.
Most cells in the human body contain the same DNA, yet different types of cells have vastly different shapes, sizes, and functions. How do these differences arise? Chemical modifications to DNA and DNA-associated proteins, called epigenetic modifications, help instruct a cell to express only a sub-set of genes, giving rise to different characteristics for different cell types. Epigenetic regulation of gene expression changes during development, and can also change as a result of environmental exposures, pharmaceuticals, aging, and diet. Some epigenetic changes promote health and normal development, while others may contribute to a variety of diseases. Three recent publications in the journal Cell from the Epigenomics program’s Reference Epigenome Mapping Centers reveal important insights about epigenomic changes that take place during development, as non-specialized stem cells differentiate into specific cell types, such as heart, brain, skin, and many more.
Dr. Bing Ren at the San Diego Epigenome Center examined epigenetic events that occur during early embryonic development, as stem cells begin to differentiate into specific cell lineages. Dr. Ren’s work shows that distinct epigenetic mechanisms regulate early and late stages of stem cell differentiation. Interestingly, several gene families that are known to play important roles in development were notably lacking in one type of epigenetic mark, called DNA methylation, in early stages of development. Some of these same genes were found to have excess levels of DNA methylation in cancer, suggesting a possible role for epigenetic regulation of developmental genes in several types of cancer.
An additional study by Drs. Bradley Bernstein and Alexander Meissner, from the Reference Epigenome Mapping Center at the Broad Institute, examined epigenomic changes that occur as human embryonic stem cells differentiate into the three germ layers that develop in an embryo: ectoderm (which becomes epidermis, nervous system, eyes, and ears), mesoderm (which becomes muscle, bone, cartilage, the circulatory system, and the urogenital system), and endoderm (which becomes parts of the gastrointestinal tract, the liver, the pancreas, and the lungs). This study revealed several discrete events that occur during differentiation into each germ layer, providing new insight into how human germ layers are specified during development. Additionally, this information may prove useful to scientists who seek to differentiate induced pluripotent stem cells (iPSCs) for the purpose of repairing or replacing a wide range of tissues damaged by disease or injury.
In a separate study, Drs. Bernstein and Meissner, along with colleagues across the Epigenomics Mapping Consortium, systematically mapped global changes in chromatin, the physical structure of DNA and proteins inside a cell. The conformation of chromatin is regulated by epigenetic factors, leading to changes in gene expression (see “A Scientific Illustration of How Epigenetic Mechanisms Can Affect Health”). By generating over 300 chromatin state maps from diverse human tissues and stem cells, the researchers have discovered signature patterns of “active” chromatin, representing genes that are being expressed, versus “repressed” chromatin, representing genes that are not expressed. During development, chromatin changes from a largely accessible state to a more restrictive state. The chromatin state maps reveal that cells of different developmental stages, or exposed to different environmental conditions, can be distinguished by characteristic differences in chromatin state maps. Prior to this study, much of what scientists knew about chromatin states came from studying cell lines derived from various model organisms.
Collectively, these studies provide a wealth of information about epigenetic dynamics in human cells within different tissues, during various developmental stages, and under a variety of environmental conditions. The extensive data sets available in these publications will be a valuable resource for researchers in a wide range of biomedical fields.
References:
From Dr. Bing Ren:
Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, et al. Epigenomic Analysis of Multi-lineage Differentiation of Human Embryonic Stem Cells. Cell, 2013 May. 153(5):1134-48. PMID: 23664764.
From Drs. Bernstein and Meissner:
Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghev J, et al. Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell, 2013 May 7. 153(5) 1149-63. PMID: 23664763.
Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, et al. Genome-wide Chromatin State Transitions Associated with Developmental and Environmental Cues. Cell, 2013 Jan. 152(3): 1-13. PMID: 23333102.
NIH Common Fund researchers link genetic variants and gene regulation in many common diseases
 Researchers supported by the NIH Common Fund have discovered that genetic differences linked to a wide variety of diseases influence how genes are turned on, or expressed. Many genetic differences, or variants, that are associated with disease do not fall within genes themselves, but are in stretches of DNA between genes, called non-coding DNA. For many years, scientists were unsure whether or not non-coding DNA served any purpose in the cell, or what the purpose could be. It is now known that these non-coding regions have important roles in regulating gene expression, but linking genetic variation in these regions with disease risk has been challenging. Dr. John Stamatoyannopolous M.D., and colleagues, funded in part by the Common Fund’s Epigenomics program, report that the majority of genetic variants linked to risk for a number of common diseases are located in non-coding DNA regions that regulate gene expression, providing new insight into how, when, and why many diseases occur. Their findings are published in the Sept. 5 online issue of the journal Science.
Researchers supported by the NIH Common Fund have discovered that genetic differences linked to a wide variety of diseases influence how genes are turned on, or expressed. Many genetic differences, or variants, that are associated with disease do not fall within genes themselves, but are in stretches of DNA between genes, called non-coding DNA. For many years, scientists were unsure whether or not non-coding DNA served any purpose in the cell, or what the purpose could be. It is now known that these non-coding regions have important roles in regulating gene expression, but linking genetic variation in these regions with disease risk has been challenging. Dr. John Stamatoyannopolous M.D., and colleagues, funded in part by the Common Fund’s Epigenomics program, report that the majority of genetic variants linked to risk for a number of common diseases are located in non-coding DNA regions that regulate gene expression, providing new insight into how, when, and why many diseases occur. Their findings are published in the Sept. 5 online issue of the journal Science.
Dr. Stamatoyannopolous and colleagues found that some of the genetic variants linked to adult-onset diseases lie in regions of DNA that regulate genes during the early stages of development, providing a potential mechanism to explain the observation that some environmental exposures in utero or during early childhood are known to increase risk of diseases that produce symptoms years or even decades later. The researchers were also able to link genetic variants in non-coding regions with the genes they regulate, which has been a major challenge in genetic studies because the genes are often located a great distance away. In addition, researchers were able to pinpoint which cell types are affected by different diseases. These results provide new insight into disease mechanisms, and suggest novel targets for therapeutics development and disease prevention strategies.
Read the University of Washington news release
Reference: Humbert R, Maurano MT, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science, 2012 Sept. 338(6099): 1190-5. PMID: 22955828.
Epigenomics researchers uncover new chemical modifications on DNA associated proteins

Epigenetic marks are chemical modifications to the genome that regulate which genes are active and which proteins are made in a cell. These marks are found on DNA as well as on the histone proteins that DNA is wrapped around. Epigenetic marks help regulate the expression of genes involved in cell development and function, and are also implicated in a growing number of diseases such as cancer, diabetes, autoimmune diseases, and mental illness (see “A Scientific Illustration of How Epigenetic Mechanisms Can Affect Health”). Drs. Yingming Zhao and Bing Ren, supported in part by the Common Fund’s Epigenomics program, along with their colleagues, have expanded our understanding of epigenetics by identifying a wealth of novel histone modification sites, as well as histone modifications that have never been described before. Using a combination of approaches in the most thorough examination of histones to date, the researchers identified 67 new histone modifications, increasing the number of known histone marks by about 70%. Some of these newly discovered histone marks correspond to types of chemical modifications that had already been described in other regions of histone proteins, but others represent an entirely new type of chemical modification of histones. One such novel modification, lysine crotonylation or Kcr, was found to label regions of the genome that are actively making proteins. In particular, Kcr modifications were found associated with genes that are activated in the testes of male mice at a specific time during development, suggesting that Kcr may regulate genes that are important for aspects of sperm cell maturation and function. The discovery of these new histone modifications expands our understanding of epigenomics, and opens the door to further research into the epigenome that regulates health and disease.
Reference: Tan M, Luo H, Lee S, Jin F, Soo Yang J, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, and Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell September 2011. 146(6): 1016-28. PMID: 21925322.
Embryonic And Induced-Pluripotent Stem Cells Reveal Own Molecular Signature

New discoveries in stem cell biology are fueling the development of new cell-based therapies for diseases such as Parkinson’s and diabetes where tissues may become diseased or damaged. Before this potential can be reached, an important, yet unanswered question is whether adult cells that are “induced” to become like embryonic stem cells – so called induced pluripotent stem cells (iPS cells) -- are actually equivalent to embryonic stem cells and can be used in cell-based therapies. Researchers in the Common Fund’s Epigenomics program are tackling this question. Read more of the story...
Reference: Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, Ecker JR. Hotspots of aberrant epigenomics reprogramming in human induced pluripotent stem cells. Nature 2011 Mar. 471(7336): 68-73. PMID: 21289626.
Mapping the Human Epigenome: A Community Resource
 Epigenetic modifications are chemical modifications to the genome that play a role in development, aging, health, and disease, and are therefore targets for therapeutic interventions. The Reference Epigenomic Mapping Consortium, funded through the Common Fund’s Roadmap Epigenomics Program, is generating genome-wide epigenomic maps for a variety of cell types. The majority of the reference epigenomes generated will contain information on epigenetic modifications having to do with a core set of histone marks, DNA methylation, and chromatin accessibility, in addition to gene expression data associating gene activity with the epigenetic modifications. A subset of reference epigenomes will also contain an expanded set of at least twenty additional histone modifications.
Epigenetic modifications are chemical modifications to the genome that play a role in development, aging, health, and disease, and are therefore targets for therapeutic interventions. The Reference Epigenomic Mapping Consortium, funded through the Common Fund’s Roadmap Epigenomics Program, is generating genome-wide epigenomic maps for a variety of cell types. The majority of the reference epigenomes generated will contain information on epigenetic modifications having to do with a core set of histone marks, DNA methylation, and chromatin accessibility, in addition to gene expression data associating gene activity with the epigenetic modifications. A subset of reference epigenomes will also contain an expanded set of at least twenty additional histone modifications.
The program’s website provides information about the program, protocols, information about data standards, and links to a variety of sites where the epigenomic data can be visualized in a genome browser or downloaded for subsequent analysis.
What Drives Our Genes? Researchers Map The First Complete Human Epigenome
 Stem cells offer enormous potential for repairing damaged tissue but historically they have been hard to obtain. Recent discoveries have shown that normal skin cells can be induced to form stem cells. This provides a readily available source of stem cells, but it’s not known if these “induced” stem cells are really equivalent to embryonic stem cells, or if the range of adult cell types made from them are normal and could be used for therapeutic purposes. An important step to answer these questions is the development of “fingerprints” of all cell types. Chemical modifications to DNA occur in different patterns in each type of cell. These modifications serve as one type of molecular fingerprint that defines what makes a liver cell a liver cell vs. a heart cell vs. a neuron vs. a “pluripotent” stem cell that has the potential to become any one of these cell types and more. To understand how an embryonic stem cell differentiates to become any type of cell in the body, we need to decipher its molecular fingerprint. We also need to know if induced stem cells have the same molecular fingerprint as embryonic stem cells.
Stem cells offer enormous potential for repairing damaged tissue but historically they have been hard to obtain. Recent discoveries have shown that normal skin cells can be induced to form stem cells. This provides a readily available source of stem cells, but it’s not known if these “induced” stem cells are really equivalent to embryonic stem cells, or if the range of adult cell types made from them are normal and could be used for therapeutic purposes. An important step to answer these questions is the development of “fingerprints” of all cell types. Chemical modifications to DNA occur in different patterns in each type of cell. These modifications serve as one type of molecular fingerprint that defines what makes a liver cell a liver cell vs. a heart cell vs. a neuron vs. a “pluripotent” stem cell that has the potential to become any one of these cell types and more. To understand how an embryonic stem cell differentiates to become any type of cell in the body, we need to decipher its molecular fingerprint. We also need to know if induced stem cells have the same molecular fingerprint as embryonic stem cells.
Researchers in the Common Fund’s Epigenomics Program have taken the first step toward this goal. They have determined a high resolution fingerprint of one type of chemical group on the DNA of human embryonic stem cells and have compared it to what is found in fibroblasts, a type of cell found in many tissue types, including skin. They found that the fingerprints varied drastically between the two cell types. In addition, an analysis of limited regions of DNA from induced stem cells yielded a partial fingerprint that showed the same characteristics as in human embryonic stem cells. This discovery yields fundamental knowledge about stem cells and indicates that induced stem cells are molecularly similar to embryonic stem cells. It provides a method to identify cells as stem cells, and it is important for future work in which these cells will be used to regenerate adult tissues.
Reference: Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009 Nov. 462(7271):315-22. PMID: 19829295.
The completed human genome sequence has been metaphorically described as “the book of life.” Expanding upon this metaphor, the map of the epigenetic DNA methylation modifications that adorn the human genome in one cell may be regarded as a single volume in the vast encyclopedia of epigenomes that may be found within the human body. The volume cover depicts a mosaic of an anatomical drawing of a human torso taken from the book “De humani corporis fabrica” (On the Structure of the Human Body) by Andreas Vesalius (1514–1564), who is often regarded as the founder of modern human anatomy. The mosaic is composed of the letter C, which represents the methylcytosine bases identified through shotgun sequencing of bisulfite-converted human genomic DNA, in which only methylated cytosines were not converted to uracil. Together this forms a graphic portrayal of the first comprehensive DNA methylomes of humans, constituting the first two volumes of the potentially vast “Encyclopedia Epigenetica”. Cover image by Ryan Lister. Letter C images: Leo Reynolds, chrisinplymouth, Karyn Christner, Eva Ekeblad (www.flickr.com). Karyotype image: NHGRI Talking Glossary of Genetics.
